Meta AI releases a protein database with the structure of hundreds of millions of proteins. It is powered by a new AI model that computes significantly faster than Deepmind's AlphaFold.
In December 2020, Deepmind unveiled AlphaFold, an AI system for predicting protein folds. The AI system is so much faster than alternative methods that it is considered by some scientists to be a solution to the nearly 50-year-old protein folding problem. In July 2021, Deepmind released AlphaFold 2 as open source and a comprehensive protein database.
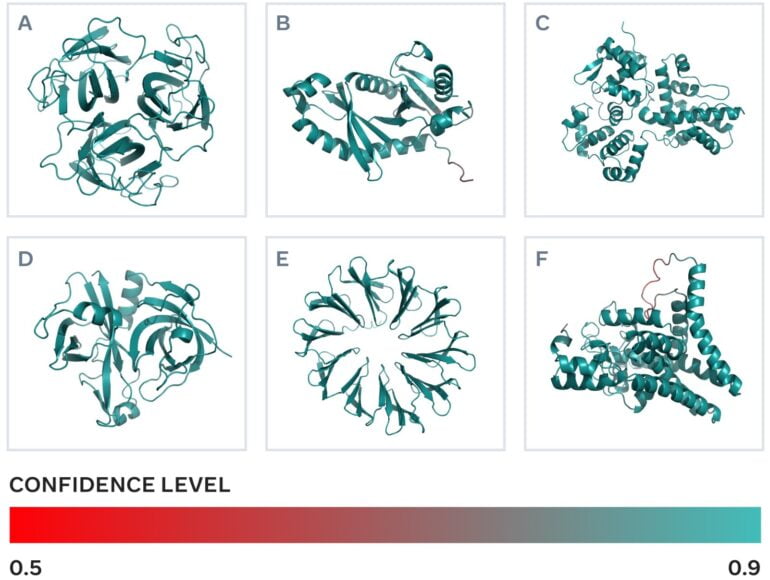
Now, researchers at Meta are also demonstrating their progress in using AI models to predict protein structures.
ESM Metagenomic Alta's database includes hundreds of millions of structure predictions
The ESM Metagenomic Atlas published by Meta includes structure predictions for 617 million proteins found in microbes in soil, the ocean, or the human body. The number of such proteins far exceeds those found in animal and plant life. Yet they are among the least understood proteins.
“These are the structures we know the least about. These are incredibly mysterious proteins. I think they offer the potential for great insight into biology,” says Alexander Rives, the research lead for Meta AI’s protein team.
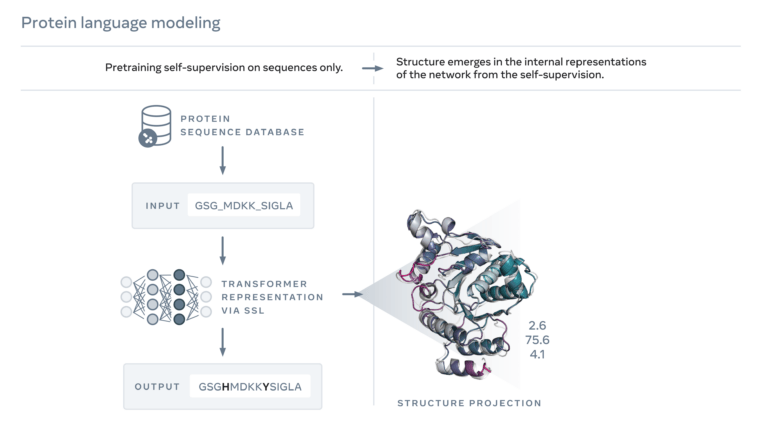
The structure predictions come from Meta's ESMFold, an AI model that relies at its core on a large language model trained with amino acid sequences of known proteins. ESMFold can complete sequences after training and predict their structures in a second step.
Meta's ESMFold is less accurate than AlphaFold, but significantly faster
According to Meta, ESMFold does not achieve the accuracy of Deepmind's AlphaFold, but it is 60 times faster. This makes Meta's approach much easier to scale to large databases, as in the case of the now-published database of metagenomic DNA. The vast majority of entries in the database come from organisms that have never been studied in the lab.
Announcing the ESM Metagenomic Atlas - the first comprehensive view of the 'dark matter' of the protein universe. Made possible by ESMFold, a new breakthrough model for protein folding from Meta AI.
More in our new blog ➡️ https://t.co/LsUhSjzqCf
1/3 pic.twitter.com/5lq48rPv5A
- Meta AI (@MetaAI) November 1, 2022
The 617 million predictions took Meta's ESMFold two weeks to complete. The model rated one-third of the predictions as high quality. In these cases, researchers can assume that the protein shape is correct and, in some cases, finer atomic-level details are discernible.

Meta is publishing the ESMFold models and a pre-print paper in addition to the ESM Metagenomic Atlas.